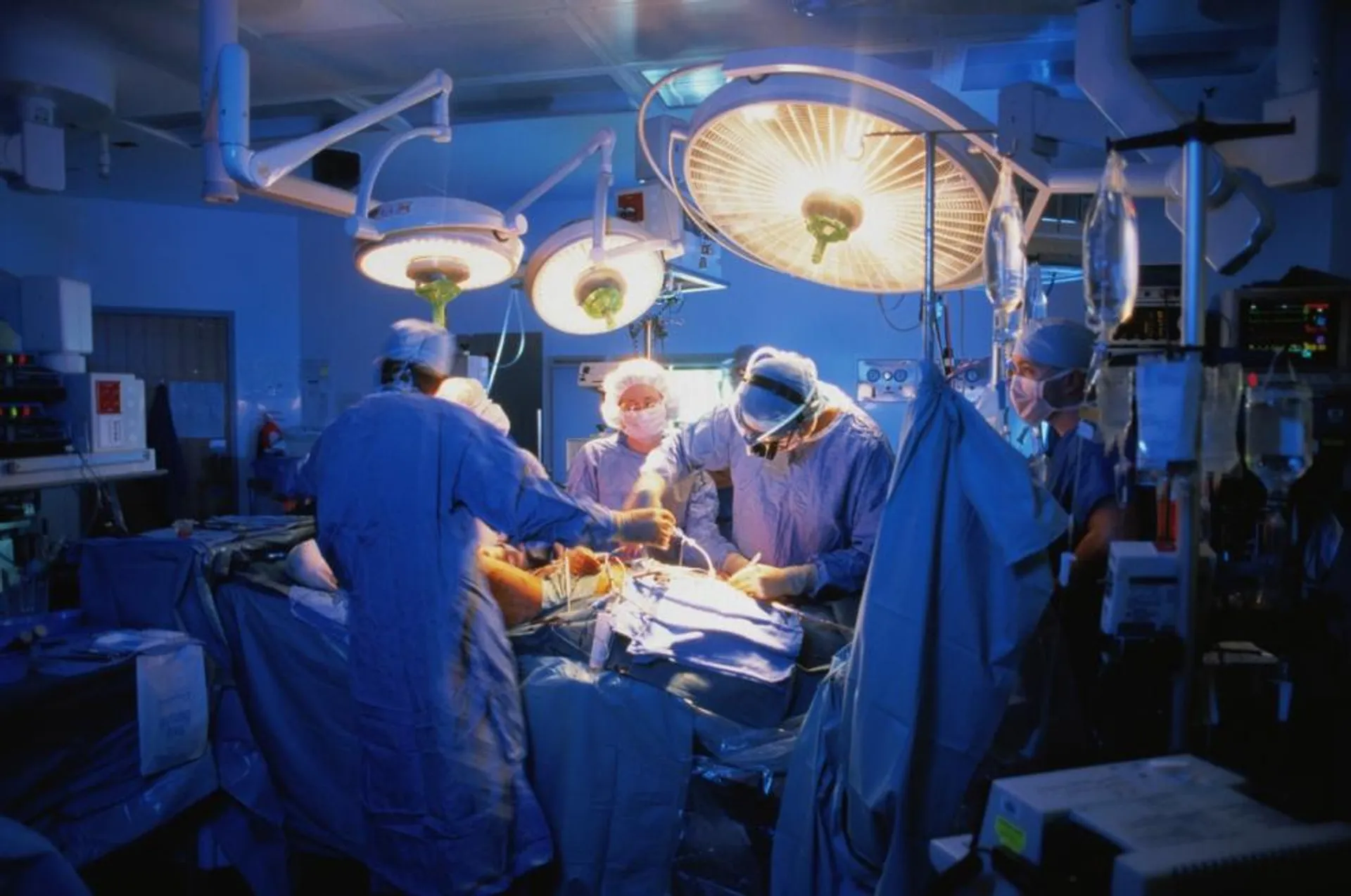
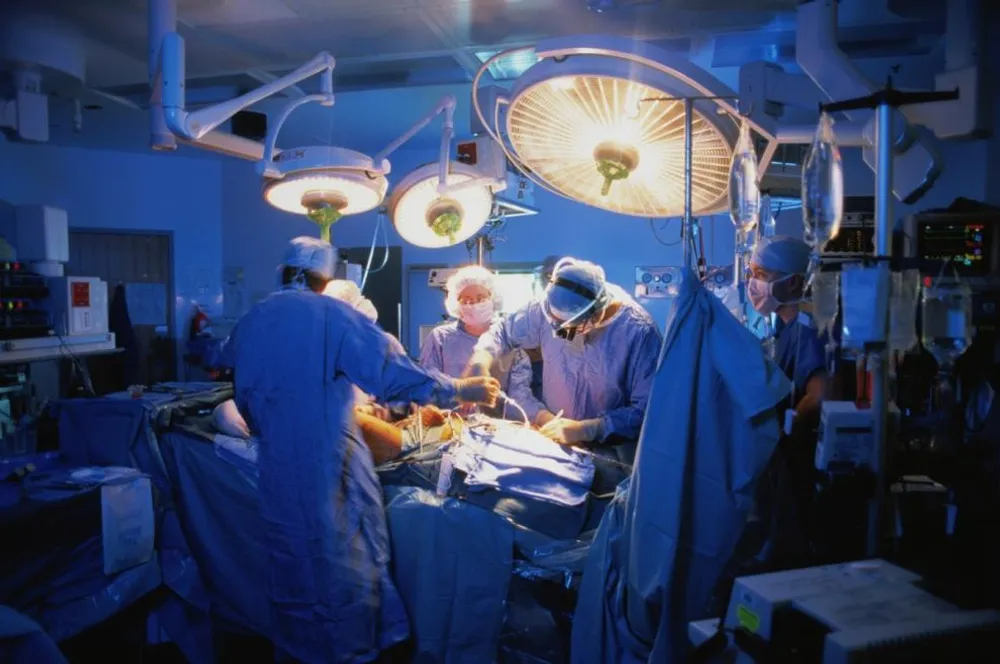
Valvole cardiache, pacemaker, protesi ortopediche e mille altre altre tecnologie mediche. Sono una moltitudine i prodotti dell’industria sanitaria (medical device) che vengono impiantati nel corpo dei pazienti. Dovrebbero essere sicuri, efficaci, solidi, sottoposti a rigorosi controlli pubblici e verificati da studi clinici di livello scientifico.
L'inchiesta giornalistica mondiale Implant Files del consorzio Icij, a cui hanno partecipato per l’Italia L’Espresso e Report, documenta invece una situazione ad altissimo rischio: una specie di Far West della salute. Un business da 350 miliardi di euro all'anno che è totalmente fuori controllo.
In Europa le aziende produttrici sfuggono legalmente alle verifiche preventive delle autorità pubbliche: devono ottenere solo una certificazione industriale da una società privata che è la stessa impresa interessata a scegliere e pagare. Ma anche negli Stati Uniti le pressioni delle lobby stanno indebolendo i controlli pubblici sulle multinazionali che inondano il mercato sanitario di congegni sempre più sofisticati e pericolosi.
«A differenza di quanto accade per i farmaci, per i dispositivi medici non è richiesta un’autorizzazione preventiva di un’autorità pubblica per l’immissione in commercio: è necessaria solo la marcatura CE, che è di competenza esclusiva del fabbricante». A confermare che queste sono le regole europee, in vigore anche in Italia dagli anni Novanta, è Marcella Marletta, direttore del dipartimento competente del nostro ministero della Salute.
La differenza con i farmaci è vistosa e allarmante. L’industria dei medicinali, prima di poter vendere un nuovo prodotto, deve dimostrare alle autorità italiane ed europee che il farmaco proposto è sicuro ed efficace: per provarlo, è obbligata a presentare diversi studi clinici di livello scientifico, condotti per anni su centinaia o su migliaia di pazienti. I dati sugli effetti collaterali sono trasparenti, controllabili dalle apposite agenzie pubbliche (come l’Ema nella Ue e la Fda negli Usa) e verificabili da scienziati indipendenti con successive sperimentazioni.
Per i dispositivi medici, invece, il mercato è liberista: il fabbricante deve ottenere solo il marchio CE da una società privata. Solo in Italia si contano undici di queste aziende di certificazione, accreditate come «organismi notificati», in concorrenza con decine di analoghe società europee. In questo modo il controllato si sceglie il controllore e lo paga quanto vuole. E se non ottiene il marchio, può cambiare certificatore.
La mancanza di controlli pubblici è criticata da vari scienziati. Enrico Gianluca Caiani, professore di bio-ingegneria al Politecnico di Milano, ha pubblicato uno studio clinico, con il luminare americano Alan Fraser, per denunciare «la necessità di maggior trasparenza per i dispositivi medici in Europa». «L’obiettivo della nostra pubblicazione - spiega il professore - è convincere la Commissione europea a varare un sistema di verifica preventiva, come avviene negli Stati Uniti». Lo studio, pubblicato l’estate scorsa sulla prestigiosa rivista The Lancet, riguarda ben 309 dispositivi cardiovascolari, ortopedici e neurologici, analizzati dal 2005 al 2010: quelli registrati in Europa hanno avuto il doppio dei problemi di sicurezza rispetto agli Stati Uniti (27 per cento contro 14). Mentre i prodotti europei ritirati dal mercato sono addirittura il triplo. «In Europa approvare un nuovo dispositivo è più facile e la procedura è più veloce», riassume il docente. «Negli Stati Uniti di regola è più lunga e severa, con una media di circa un terzo di bocciature». Una diversità di regole che nel 2011 portò l’allora capo della Fda americana, Jeffrey Shuren, a dichiarare: «Nel sistema europeo i pazienti vengono utilizzati come cavie».

La certificazione privata europea può fondarsi su un unico studio clinico effettuato su poche decine di persone, che resta riservato: un segreto industriale, custodito dal produttore e dal suo consulente-controllore. E quando uno dei tanti certificatori dà via libera, il dispositivo può essere impiantato su tutti i pazienti dell’Unione europea. E in molte altre nazioni, dall’Asia al Sudamerica, che si fidano del marchio CE.
In Italia, tra le società di certificazione, c’è una sola struttura pubblica: l’Istituto superiore di sanità, il braccio scientifico del ministero della salute. Un’istituzione creata per tutelare l’interesse pubblico, a differenza delle società private che hanno finalità di profitto. Il problema è che cinque dirigenti dell’ente statale, in carica dal 2010 al 2014, sono stati rinviati a giudizio per falso. Il processo si è aperto il 7 novembre scorso nel tribunale di Roma. Il caso giudiziario nasce da uno scoop di Report. Nell’ottobre 2014 i giornalisti d’inchiesta della Rai riescono a entrare nei laboratori dell’istituto. E scoprono che i macchinari per le prove tecniche non funzionano o non esistono.
L’indomani, la procura di Roma manda al ministero la Guardia di Finanza, che conferma: gli ultimi veri test di laboratorio risultano eseguiti nel 2009; da allora i macchinari, già obsoleti, sono rimasti inutilizzati, impolverati, fermi da almeno un quinquennio. Di qui l’accusa ai manager dell’istituto pubblico di aver rilasciato «false certificazioni», senza fare alcun controllo, per oltre 80 «dispositivi ad alto rischio: pacemaker, neurostimolatori e stent coronarici», tutti prodotti da aziende italiane. Gli imputati si difendono e negano qualsiasi illecito. Il processo proseguirà in gennaio. Nell’attesa, un commissario ministeriale, in una relazione, ha denunciato due ricercatori sospettati di aver favorito «l’accesso abusivo» dei colleghi di Report.
Dubbi pesantissimi riguardano anche i certificatori privati. L’inchiesta del consorzio nasce dal lavoro di una giornalista della televisione pubblica olandese sui dispositivi-barriera contro il prolasso degli organi riproduttivi, che può verificarsi dopo un parto complesso. I più diffusi somigliano a reti di plastica, applicate tra l’utero e la vescica, che si saldano al tessuto organico. Molti hanno effetti terribili: circa centomila donne hanno già intentato cause di risarcimento. Quindi la collega, Jet Schouten, ha comprato al supermercato una confezione di mandarini, ha tagliato la rete rossa che li conteneva e ha chiesto a tre società private di certificarla, allegando uno studio clinico vistosamente falso. Con dati negativi da film dell’orrore: nei test (inventati) quel dispositivo rendeva invalide un terzo delle donne. Ciò nonostante, i manager delle tre società europee, tra cui un’azienda italiana, sono stati videoregistrati mentre assicuravano che, in cambio di una lauta parcella, erano pronti a concedere il marchio CE. Alla rete per i mandarini.

L’anno scorso l’Unione europea ha varato un nuovo regolamento sui dispositivi medici, che entrerà in vigore gradualmente entro il 2020. Quando iniziò l’iter della riforma, dopo i primi scandali, l’obiettivo dichiarato era la creazione di un’autorità pubblica, come negli Usa. Gli atti ottenuti dal consorzio documentano continue pressioni delle lobby industriali. Che sul punto centrale hanno vinto: il sistema europeo resterà fondato sulle certificazioni private.


